
Computational Nanomechanics of Materials (former KIT Young Investigator Group)
In materials science of structural materials the state of microstructure determines the materials behavior in processing (production) and during materials usage in devices. Additionally, the microstructures evolve dynamically as a results of defect nucleation, defect motion, and/or defect interaction. The group “Computational Nanomechanics of Materials” investigates materials behavior with computer simulations as inspiration for synchronized experiments. Central to us is the dynamics of extended defects in these dynamic microstructures; or put simple: “The mechanics of microstructures.”
Our approach
Our approach to materials behavior by computational materials science includes an interface to experiments to enable complementary insights and to utilize simulations as a predictive inspiration for novel experiments.
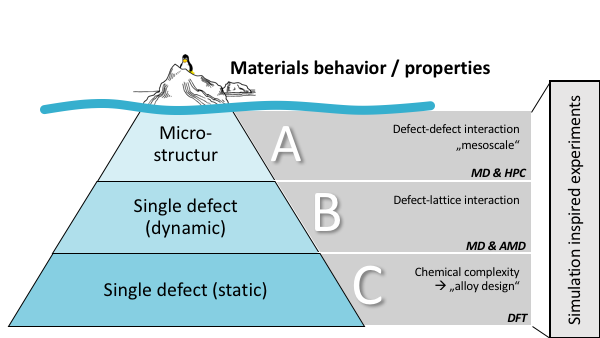
| (A) | On microstructure level (mesoscale), we use molecular dynamics (MD) simulations on high performance computers (HPC) to simulate the dynamics of complete microstructure with atomistic resolution. |
| (B) | For the dynamics of single defects the focus is on the thermodynamics and kinetics of defect nucleation and defect motion. This also include sophisticated models and simulations methods to explore the rare-event dynamics of thermally activated processes to reach with atomistic fidelity time scales, which approach experimentally relevant regimes. |
| (C) | With the focus on static properties of defects in chemical different environments with ab-initio (Density Functional Theory) simulations methods we predict alloying trends for the properties related defect dynamics. |
Research topics
 |
Impurities and plastic deformation in bcc metals Methods: MD, DFT and Nanoindentation |
 |
Deformation behavior and mechanisms in defect-scarce Au nanowires Methods: MD, in-situ SEM, TEM |
 |
Grain boundary motion in extreme conditions and in nanomaterials Methods: MD, TEM, shock loading |
 |
Plastic deformation of nanocrystalline metals Methods: MD, in-situ diffraction, in-situ ACOM STEM |